
Article title: Syndrome VEXAS comme nouveau diagnostic différentiel de la spondyloarthrite du sujet âgé
First author: Jain H
Journal: MEDITERRANEAN JOURNAL OF RHEUMATOLOGY
Link to the article: https://pubmed.ncbi.nlm.nih.gov/39463879/
Author of the abstract: Philippe Mertz

VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) is an autoinflammatory disease associated with somatic mutations in the UBA1 gene, mainly observed in people over the age of 50. This gene encodes a key enzyme in the ubiquitination process, whose dysfunction leads to the activation of several inflammatory pathways. Signs suggestive of VEXAS syndrome include macrocytic anemia, neutrophilic dermatoses (such as Sweet's syndrome), pulmonary and ophthalmological involvement, thrombosis, and various rheumatological manifestations. The latter often present as arthralgia or mono-, oligo-, or polyarthritis.
Spondyloarthritis in older individuals is a rare condition that can be axial and/or peripheral, with an inflammatory component that is often more pronounced than in younger individuals. It requires the elimination of certain specific differential diagnoses, including microcrystalline rheumatism, rhizomelic pseudo-polyarthritis, giant cell arteritis, and paraneoplastic rheumatism.
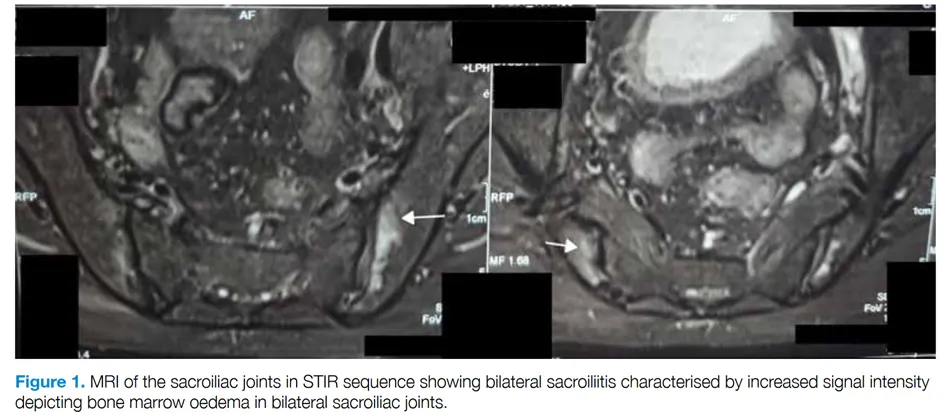
The authors report the case of a 67-year-old man living in India with peripheral polyarthritis associated with chronic inflammatory low back pain. Skin lesions had also been observed for a month, and biopsy revealed neutrophilic dermatosis, which responded well to corticosteroid therapy. Rheumatological tests showed the presence of HLA B27, active bilateral sacroiliitis on MRI, synovitis of the wrists, and bilateral retrocalcaneal bursitis. Initial treatment with sulfasalazine and NSAIDs failed, and the introduction of methotrexate at 15 mg/week was also ineffective. Further investigations revealed macrocytic anemia (Hb = 8.3 g/dL, MCV = 106 fL) associated with a biological inflammatory syndrome (CRP = 38 mg/L). These abnormalities led to Sanger sequencing of the UBA1 gene, confirming the diagnosis of VEXAS syndrome due to the p.Met41Leu mutation. Corticosteroid therapy combined with sulfasalazine at 1 mg/day controlled the joint symptoms.
Two other cases of spondyloarthritis in older patients associated with VEXAS syndrome have been reported:
A 57-year-old man living in France, presenting with a similar clinical picture, including peripheral polyarthritis, bilateral sacroiliitis, HLA-B27 positivity, and biological inflammatory syndrome (2). When the first inflammatory symptoms appeared at the age of 57 in 2010, treatment with anti-TNF alpha initially provided good control of the symptoms. Subsequently, he developed acute anterior uveitis, chondritis of the ear and nose, chronic inflammatory bowel disease (IBD), and neutrophilic dermatosis. Several lines of treatment were attempted without lasting success, including methotrexate, azathioprine, cyclophosphamide, baricitinib, infliximab, tocilizumab, anakinra, and ustekinumab. Finally, a combination treatment of intravenous immunoglobulins (2 g/kg every 4 weeks) and secukinumab (300 mg every 4 weeks) proved effective. The diagnosis of VEXAS syndrome associated with the Met41Thr mutation was made retrospectively in 2020.
A 74-year-old Caucasian man with a recent history of venous thrombosis in the lower limbs presented with inflammatory low back pain with bilateral sacroiliitis on MRI, macrocytic anemia (Hb = 8.8 g/dL and MCV = 101 fL), but without significant biological inflammatory syndrome (3). He subsequently developed progressive anemia (Hb = 8.5 g/dL and MCV = 100 fL), purpura of the lower limbs, erythema nodosum, and chondritis of the ear. He was diagnosed with VEXAS syndrome associated with the Met41Thr mutation of the UBA1 gene. After several treatments failed, including tocilizumab complicated by abscessed diverticulitis, azacitidine at the standard dose of 75 mg/m²/day led to a significant reduction in corticosteroid therapy (from 40 mg/day to 5 mg/day) and clinical improvement of symptoms.
The manifestations of VEXAS syndrome are varied and still poorly defined. It appears to be one of the new differential diagnoses to consider in cases of spondyloarthritis in older patients, particularly those aged 50 and over, given the age of onset of VEXAS syndrome and especially in the presence of a biological inflammatory syndrome, hematological abnormalities with associated anemia, particularly macrocytic anemia, or other suggestive findings such as neutrophilic dermatosis. An atypical history or the appearance of new extra-rheumatological symptoms, such as venous thrombosis, inflammatory dermatoses (particularly neutrophilic), chondritis, or pulmonary involvement, should also point to this diagnosis. In addition, resistance to initial treatment with NSAIDs may be a warning sign, especially since the hematological abnormalities associated with VEXAS syndrome may appear secondarily.
REFERENCES
Jain H, Roy D, Mavidi S, Haldar S, Mondal S, Bhattacharya P, et al. Elderly Onset Spondyloarthropathy and VEXAS Syndrome: A Case Report. Mediterr J Rheumatol. 2024 Sep;35(3):490–3.
Magnol M, Couvaras L, Degboé Y, Delabesse E, Bulai-Livideanu C, Ruyssen-Witrand A, et al. VEXAS syndrome in a patient with previous spondyloarthritis with a favourable response to intravenous immunoglobulin and anti-IL17 therapy. Rheumatology (Oxford). 2021 Sep 1;60(9):e314–5.
Pereira da Costa R, Sapinho G, Bandeira M, Infante J, Marques T, Mimoso Santos C, et al. Case report: VEXAS syndrome: an atypical indolent presentation as sacroiliitis with molecular response to azacitidine. Front Immunol. 2024;15:1403808.

