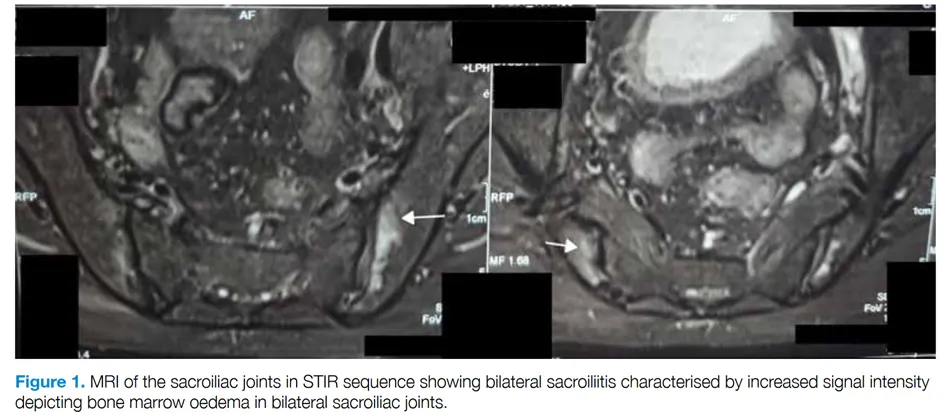
First author: Yixiang Yves-Jean Zhu
Journal: European Journal of Internal Medicine
Reference: https://doi.org/10.1016/j.ejim.2025.05.023

Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome is a rare autoinflammatory disease associated with somatic pathogenic variants in the UBA1 gene. First described in 2020, it has since been reported in many countries, but its distribution across the world remained unclear. We conducted a literature review between October 2020 and April 2025, identifying more than 670 cases across 32 countries and 4 continents. Among patients with documented origins, several ethnic groups were represented, including Caucasian, East and South Asian, Middle Eastern, and South American. These findings confirm that VEXAS syndrome diverse ethnic backgrounds and has a broad worldwide distribution. It is therefore crucial to consider VEXAS in patients with compatible symptoms, regardless of their country or ancestry, to avoid diagnostic delays.