
- Philippe Mertz
- 1 sept. 2024
- 4 min de lecture
Dernière mise à jour : 20 oct. 2025
Titre de l'article:Syndrome VEXAS comme nouveau diagnostic différentiel de la spondyloarthrite du sujet âgé
Premier auteur: Jain H
Revue: MEDITERRANEAN JOURNAL OF RHEUMATOLOGY
Lien vers l'article: https://pubmed.ncbi.nlm.nih.gov/39463879/
Auteur du résumé: Philippe Mertz

Le syndrome VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) est une maladie autoinflammatoire associée à des mutations somatiques du gène UBA1, principalement observée chez les personnes de plus de 50 ans. Ce gène code une enzyme clé du processus d'ubiquitination, dont la dysfonction entraîne l'activation de plusieurs voies inflammatoires. Les signes évocateurs du syndrome VEXAS incluent une anémie macrocytaire, des dermatoses neutrophiliques (comme le syndrome de Sweet), des atteintes pulmonaires, ophtalmologiques, des thromboses, et des manifestations rhumatologiques variées. Ces dernières se présentent souvent sous forme d'arthro-myalgies ou de mono-, oligo-, ou polyarthrites.
La spondyloarthrite du sujet âgé est une entité rare, pouvant être axiale et/ou périphérique, avec une composante inflammatoire souvent plus marquée que chez le sujet jeune. Elle impose d’éliminer certains diagnostics différentiels spécifiques, notamment les rhumatismes microcristallins, la pseudo-polyarthrite rhizomélique, l’artérite à cellules géantes, ou encore des rhumatismes paranéoplasiques.
Les auteurs rapportent le cas d’un homme de 67 ans habitant en Inde présentant une polyarthrite périphérique associée à des lombalgies chroniques d’horaire inflammatoire. Depuis un mois, des lésions cutanées avaient également été observées, et la biopsie a révélé une dermatose neutrophilique, qui a bien répondu à une corticothérapie. Le bilan rhumatologique a montré la présence de l’HLA B27, une sacro-iliite bilatérale active à l’IRM, des synovites des poignets et une bursite rétrocalcanéenne bilatérale. Le traitement initial par sulfasalazine et AINS a échoué, et l’introduction de méthotrexate à 15 mg/semaine s’est également révélée inefficace. Des explorations complémentaires ont mis en évidence une anémie macrocytaire (Hb = 8,3 g/dL, VGM = 106 fL) associée à un syndrome inflammatoire biologique (CRP = 38 mg/L). Ces anomalies ont conduit au séquençage par Sanger du gène UBA1, confirmant le diagnostic de syndrome VEXAS par la mutation p.Met41Leu. Une corticothérapie combinée à un traitement par sulfasalazine à 1 mg/jour a permis de contrôler les symptômes articulaires.
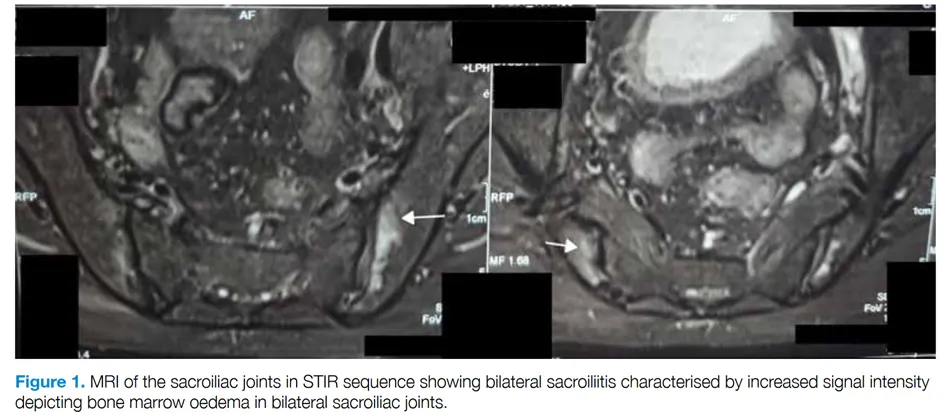
Deux autres cas de spondyloarthrite du sujet âgé associés au syndrome VEXAS ont été rapportés :
Homme de 57 ans habitant en France, présentant un tableau clinique similaire, incluant une polyarthrite périphérique, une sacro-iliite bilatérale, une positivité pour HLA-B27 et un syndrome inflammatoire biologique (2). Lors des premières manifestations inflammatoires à 57 ans en 2010, un traitement par anti-TNF alpha avait permis un bon contrôle initial des symptômes. Par la suite, il a développé une uvéite antérieure aiguë, des chondrites de l’oreille et du nez, une maladie inflammatoire chronique de l’intestin (MICI), et une dermatose neutrophilique. Plusieurs lignes thérapeutiques ont été tentées sans succès durable, incluant le méthotrexate, l’azathioprine, la cyclophosphamide, le baricitinib, l’infliximab, le tocilizumab, l’anakinra et l’ustekinumab. Finalement, un traitement combinant des immunoglobulines intraveineuses (2 g/kg toutes les 4 semaines) et le sécukinumab (300 mg toutes les 4 semaines) a montré une efficacité. Le diagnostic de syndrome VEXAS associé à la mutation Met41Thr a été posé rétrospectivement en 2020.
Homme de 74 ans d’origine caucasienne avec antécédent de thrombose veineuse des membres inférieurs récent, présentait des lombalgies inflammatoires avec une sacro-iliite bilatérale à l’IRM, une anémie macrocytaire (Hb = 8,8 g/dL et VGM = 101 fL), mais sans syndrome inflammatoire biologique significatif (3). Par la suite, il a développé une progression de l’anémie (Hb = 8,5 g/dL et VGM = 100 fL), un purpura des membres inférieurs, un érythème noueux et une chondrite de l’oreille. Le diagnostic de syndrome VEXAS associé à la mutation Met41Thr du gène UBA1 a été posé. Après l’échec de plusieurs traitements, notamment le tocilizumab compliqué d’une diverticulite abcédée, l’azacitidine à la dose standard de 75 mg/m²/jour a permis une réduction significative de la corticothérapie (de 40 mg/jour à 5 mg/jour) et une amélioration clinique des symptômes.
Les manifestations du syndrome VEXAS sont variées et encore mal définies. Il semble être un des nouveaux diagnostics différentiels à envisager en cas de spondyloarthrite du sujet âgé, notamment à partir de 50 ans vu l’âge de survenue du syndrome VEXAS et en particulier en présence d’un syndrome inflammatoire biologique, d’anomalies hématologiques avec anémie en particulier macrocytaire associées ou d’autres atteintes évocatrices comme une dermatose neutrophilique. Des antécédents atypiques ou l’apparition de nouveaux symptômes extra-rhumatologiques, tels que des thromboses veineuses, des dermatoses inflammatoires (notamment neutrophiliques), des chondrites ou des atteintes pulmonaires, doivent également orienter vers ce diagnostic. De plus, une résistance au traitement initial par AINS peut constituer un signal d’alerte, d’autant que les anomalies hématologiques associées au syndrome VEXAS peuvent apparaître secondairement.
REFERENCES
Jain H, Roy D, Mavidi S, Haldar S, Mondal S, Bhattacharya P, et al. Elderly Onset Spondyloarthropathy and VEXAS Syndrome: A Case Report. Mediterr J Rheumatol. 2024 Sep;35(3):490–3.
Magnol M, Couvaras L, Degboé Y, Delabesse E, Bulai-Livideanu C, Ruyssen-Witrand A, et al. VEXAS syndrome in a patient with previous spondyloarthritis with a favourable response to intravenous immunoglobulin and anti-IL17 therapy. Rheumatology (Oxford). 2021 Sep 1;60(9):e314–5.
Pereira da Costa R, Sapinho G, Bandeira M, Infante J, Marques T, Mimoso Santos C, et al. Case report: VEXAS syndrome: an atypical indolent presentation as sacroiliitis with molecular response to azacitidine. Front Immunol. 2024;15:1403808.


